
3. A guideline for analysis of dyes in plant and textile extracts
3. A guideline for analysis of dyes in plant and textile extracts
3.1 Mechanism of dye binding (for plant/animal-based dyes)
For the purposes of this discussion, there are only three types of dyes: mordant dyes, vat dyes and direct dyes. Before the advent of synthetic fibers (nylon in 1935) the only types of fibers widely used for textiles were proteinaceous (silk and wool) and cellulosic (cotton, ramie, hemp, etc.; and, beginning in the late 1890s, rayon, which is regenerated cellulose).
3.1.1 Mordant dyes. The most easily dyed fibers are the proteins, silk and wool, which are by far the most frequently seen fibers in historical textiles and rugs. All proteins contain negatively charged carboxylate groups (-COO-) due to the side chains of aspartic and glutamic acids, and positively charged amino groups (-NH3+) due to lysine and arginine. Dyes bind to these charged groups. However, most natural dyes are essentially neutral and require a mordant to give them a charge. Cellulose (e.g., cotton) contains essentially no ionic groups and therefore is hard to dye.
The most common mordants are trivalent cations, e.g., Al3+, Fe3+, Cr3+, though some others, Cu2+, Sn4+, have also been used. By far, the most commonly used mordant ion is Al3+. These cations form charged coordination complexes with neutral dye molecules, which act to form bridges between the dye molecules and the proteins ( Figure 3.1).
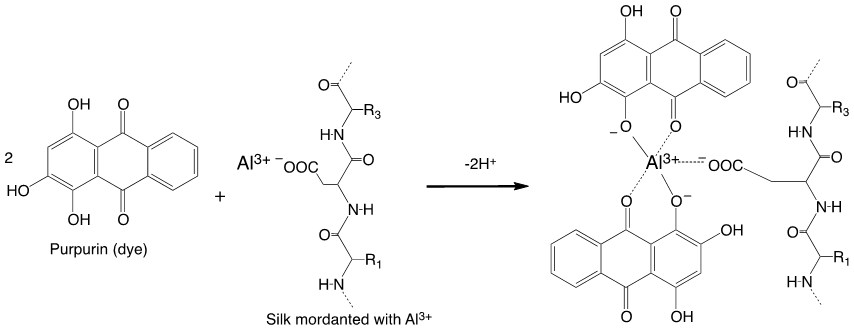
Figure 3.1. Binding of a neutral dye (purpurin) via a mordant (Al3+) to carboxylate groups in silk.
The most common (or perhaps only) complexing structures in natural dyes are the 3-hydroxy-1-ketone structure seen, for example, in Figures 3.1 and 3.2 for purpurin, and the 1,2-diphenol seen in tannins and also in many flavonoids and anthraquinones (Figure 3.2).

Figure 3.2. Complexation structures in mordant dyes.
3.1.2 Vat dyes. Vat dyes depend on their extreme insolubility in water, and essentially precipitate on the surfaces of or in the interstices within the fibers. No mordant is needed for these compounds. The most common of these types are indigo and its brominated relatives (Tyrian purple) and carthamin (safflower red). No complexing agent is needed for extraction—only a good extraction solvent, e.g., dimethylsulfoxide or pyridine. Water in either of these solvents decreases the solubility.
3.1.3 Direct dyes. A direct dye is one that can be used without a mordant. These are of two types: ionic and non-ionic.
Ionic direct dyes contain a strong cationic group (in basic dyes), such as a quaternary amine, or a strong anionic group (in acid dyes), most often a sulfonic acid group. As these dyes contain essentially permanent charges, they will bind to ionic groups in silk and wool (see section 3.1.1 above) without the need for a mordant.
Of the natural dyes, only the protoberberines, found in species of cork tree, barberry and some other plants, are direct dyes, due to the presence of positively charged quaternary ammonium groups. However, a number of the early synthetic dyes are direct dyes, namely, the aminotriphenylmethine (cationic, or basic) types, as well as many azo-dyes containing sulfonic acid groups (anionic, or acidic).
The most common non-ionic direct dyes are the carotinoid dyes (from crocus, gardenia and annatto), which are neutral substances, usually crocetin and related compounds, that bind to silk and wool without the need for a mordant. It is well known to protein chemists that proteins contain hydrophobic domains, so presumably these dyes bind via hydrophobic interaction with these domains in proteins, e.g., wool and silk. It can be predicted that these interactions will be rather weak, so probably carotinoid-dyed textiles are not very fast to washing. In fact, after analyzing hundreds of textile specimens from around the world, we have never encountered a carotinoid dye.
3.2 Principles of extraction of dyes from textiles and other materials
Most natural dyes owe their washing fastness to mordant mediated complexation with ionic groups in silk or wool. In order to be able to extract the dye molecule from the fiber, this complex must be destroyed. The role of a chelation agent or strong acid in the extraction of these dyes from silk or wool is to neutralize the dye molecule in the complex so that it can be extracted out with water and/or some organic solvent (Figure 3.3). If an acid is used to break up the complex, a strong acid should be used so as to give the extraction a pH that is lower than the pKa of the silk (or wool) carboxyl groups (~4.5). This can be accomplished with HCl or trifluoroacetic acid. Formic acid (pKa = 3.75) is a relatively strong carboxylic acid but is not as strong or effective as HCl or trifluoroacetic acid—but it works if the concentration is high enough. The problem with strong acids is that they can (and often do) catalyze hydrolysis of glycosidic bonds, which are usually present in flavonoid dyes.

Figure 3.3. Disruption of the dyed silk complex using a good chelator (oxalic acid; (COOH)2) or a strong acid (e.g., HCl, trifluoroacetic acid or even formic acid).
3.2.1 Choice of a chelation agent. Many chelators (see Table 3.1) of polyvalent metals are known, but we favor oxalic acid (or oxalate ion) [P. Guinot and C. Andary (2006), “Molecules involved in the dyeing process with flavonoids.” Oral presentation, 25th Meeting of Dyes in History and Archaeology, Suceava, Romania] for two reasons. First, it is a very strong chelator, and second, oxalic acid, even though it is a solid, has a relatively high vapor pressure and evaporates in the high vacuum of the mass spectrometer. In contrast, EDTA, which is also an excellent chelator, does not vaporize, as it is an internal salt, and can build up in the mass spectrometer.
Table 3.1. Chelators (complexing agents for mordant metal ions)
| Compounds | Comments |
|---|---|
| EDTA (Ethylenediamine tetraacetic acid, Na salt) | Not volatile; not recommended for MS |
| Oxalic acid | Solid, but volatile in high vacuum; usable with MS |
| Fluoride (as in HF) | Good complexer, but HF is hazardous and must be handled carefully; it etches glass. |
3.2.2 Choice of an extraction solvent. Some of the solvents commonly used for the extraction of dyes from textiles are listed in Table 3.2. All of these are miscible with water in all proportions.
One component needed for any efficient extraction mixture is water. Water is highly polar and can penetrate and cause swelling of fibers such as silk, wool and cellulose, which are themselves highly polar. Consider, too, that the dyeing of these fibers is always done in water, which presumably helps the dye molecules to diffuse into the fibers. Diffusion of all chemical processes is accelerated by heat, so being able to heat the solvent to a high temperature is beneficial. But if the solvent boiling point is low, then there is a limit on how high the sample can be heated. On the other hand, if the boiling point of a solvent component is very high, it can be difficult to remove it by evaporation.
Table 3.2. Solvents reported as having been used as components of solutions for extraction of dyes from textiles.
| Solvents | B.p. (°C) | Comments |
|---|---|---|
| Acetone | 56 | |
| Methanol | 64 | |
| Trifluoroacetic acid | 72 | |
| Water | 100 | |
| Formic acid | 101 | |
| HCl (contstant boiling) | 109 | B.p. of ~20% azeotrope with water (~5.8 M). HCl corrodes stainless steel and must be completely removed before sample is applied to the HPLC system. |
| Pyridine | 115 | Forms 40% azeotrope with water; b.p. 94 °C |
| Acetic acid | 118 | |
| Dimethylformamide | 153 | Slow to evaporate; impurities in some batches of DMF form adducts with dye molecules (+71-Da peak) |
| Dimethylsulfoxide | 189 | Excellent solvent, but very difficult to evaporate |
After trying many combinations of solvents, we have settled on a 1:1 mixture of pyridine and water. This mixture allows us to heat the extraction mixture to about 100 °C in ordinary “snap-cap” plastic microcentrifuge tubes without worrying about super-heating leading to uncontrolled expulsion (“bumping”) or evaporation of the solvent. Because of its aromaticity, pyridine is also a good solvent for the water-insoluble indigoid dyes. Because pyridine is essentially neutral (actually a very weak base), there is no possibility of cleavage of glycosidic linkages, as is seen with HCl or trifluoroacetic acid. Solvents such as acetone and methanol have low boiling points, which limits the temperatures to which they can be heated.
In the past we used dimethylformamide (DMF), which performs well, but noticed the formation of adducts of +71 Da to dye molecules, depending on the purity of the DMF (MW = 73). We never could identify this impurity, but decided to abandon DMF because of the uncertainty about purity. Dimethylsulfoxide (DMSO) is an excellent solvent for almost everything, but is difficult to evaporate because of its high boiling point. DMSO does not interfere with HPLC analysis and diode array detection, but it may not be compatible with mass spectrometric detectors (we have not tried it).
Pyridine-water may not be the “best” solvent for all dyes, but it does work fairly well for all of the dyes we have encountered. When one encounters a textile sample colored with a dye that is unknown, it is important that a fairly high proportion of any dye can be extracted and analyzed.
[Note: The vat dye, carthamin (from safflower), is unstable to heat in the presence of water (R. Laursen and C. Mouri, “Decomposition and Analysis of Carthamin in Safflower-Dyed Textiles,” ePS 10, 35-37 (2013)). We have noted, however, that it seems to be stable to extraction with non-aqueous solvents, such as 5% formic acid in methanol, which we used in the past. Therefore, it is suggested that a non-aqueous solvent be used if carthamin is suspected.]
3.2.3 Evaporation of extraction solvent. The method used to evaporate extraction solvents depends on the facilities and budget available to the analyst.
Vacuum evaporation. This is the method we have used in our work. Removal of extraction solvent by vacuum evaporation requires a somewhat complex set up, but has the advantage that many (up to about 25) extracts can be evaporated at the same time at low temperature and without attention. The set up requires (1) a vacuum desiccator in which to place the samples, (2) a trap cooled by dry ice-acetone or liquid nitrogen in which the solvent is condensed, (3) a manometer to measure the pressure, (4) (preferably) a tower filled with NaOH pellets to protect the vacuum pump from acids and (5) a mechanical vacuum pump capable of achieving a pressure of 0.1 – 1.0 mm Hg or less (a water aspirator or diaphragm pump is not adequate). Some tubing and valves are also needed (see Figure 3.4). This system can be used to evaporate dimethylformamide (b.p. 153 °C) at room temperature. [However, we do not recommend using DMF for reasons discussed above.]

Figure 3.4. Set-up for evaporation of solvents under vacuum. (A) Hose to vacuum pump, (B) sodium hydroxide tower, (C) manometer, (D) dry ice/acetone trap, (E) vacuum manifold (can be replaced by a 2-way vacuum stopcock, (F) desiccator containing samples to be evaporated.
Evaporation is done in open 1.8-mL plastic (polypropylene) microcentrifuge tubes. A piece of Parafilm is stretched across the top of the tube and is punctured with about 20 small holes using a pin or needle. The liquid in the tubes is frozen in dry ice and then the tubes are quickly placed in a rack in the desiccator and the vacuum is applied. The purpose of cooling/freezing of the samples is to prevent boiling (‘bumping”) of the liquid as the vacuum is applied. The role of the punctured Parafilm is to prevent the liquid, should it “bump,” from spraying over all the other tubes and contaminating them. At very low pressures, evaporation is primarily by diffusion, so there have to be enough holes (in the Parafilm) to allow the solvent vapors to diffuse through.
Nitrogen stream evaporation. In this method, the sample tubes are evaporated under a stream of nitrogen, which is delivered through needles whose tips are placed above the liquid. If an appropriate sample tube rack is available, it can be placed in a warm bath to speed evaporation. If a manifold is available, a number of samples can be evaporated at the same time (see Figure 3.5). Such an apparatus needs to be placed in a fume hood, as some solvents (like pyridine) are toxic. Nitrogen is usually used because it is clean, inert and less expensive than gases like argon. Air should not be used because the oxygen in it may destroy some of the dye molecules.

Figure 3.5. Set-up for nitrogen stream evaporation. (A) Sample tubes, (B) needles directing gas stream to samples, (C) gas manifold (this one holds nine needles), (D) tube to nitrogen tank
This method is particularly good for low boiling solvents, which have a tendency to “bump” when heated or subjected to a vacuum. One has to carefully regulate the gas stream flow, as too high a flow rate can blow the sample out of the tube. Also the needles have to be carefully cleaned so as to prevent cross-contamination of samples. It is best to extend the steel needles with a few centimeters of Teflon tubing, which can be removed for cleaning or replaced.
3.2.4 Weighing the sample. We have found that weighing samples of unknowns (e.g., museum specimens) does not give any useful information and therefore is not worth the effort.
Many unknowns weigh considerably less than 1 mg and cannot be weighed accurately on an ordinary analytical scale, which is usually is accurate only to about ±0.1 mg. True microbalances, which weigh microgram amounts, are rarely found these days and are hard to use. But the primary reason for not weighing the sample, beside its giving little or no useful information, is that one often has only a small amount of sample and has to use all of it and hope there is enough to see something. In our own work, we like to use 0.5 mg of yarn or thread, but sometimes have had to make do with almost invisible samples.
3.2.5 Extraction volumes. Newer HPLC systems incorporating capillary columns and more sensitive diode array and mass spectrometric detectors have a sensitivity that can be 10, or even 100, times greater than the system we have used for some years. In principle, this means that one could use 1/10 the usual sample size. However, the problem remains of manipulating the sample before it is applied to the HPLC column. With our current system, we dissolve the unknown (after extraction from the fibers and evaporation) in 50 µL of solvent (methanol-water) and inject 20 µL onto the HPLC column. With a high-sensitivity system, one could inject 2 µL and get the same signal size. This has the advantage that one can repeat the analysis if something goes wrong, but it does not reduce the size of the initial sample, because one cannot reduce the volume of the solution from 50 µL to 5 µL. It is very difficult to avoid losses when dealing with very small volumes, due to solvent evaporation and losses due to adherence to the surfaces of vials, pipette tips, filters (if used) etc. One might be able to work at the 25-µL level, but it would be difficult to work with less.
Another problem is that capillary HPLC columns, although they give superior separations, and are needed for high sensitivity analyses, tend to be more problematic with regard to clogging than, say, 2.1-mm diameter packed particle HPLC columns.
3.2.6 Removal of particulate matter. It is essential to remove particulate matter from samples before HPLC analysis, as particles can clog the HPLC column. We have always used a microcentrifuge, operated at about 12,000 rpm and designed for microcentrifuge tubes, for this purpose. Microcentrifuges are expensive, but one can often find a used one on Ebay or at a used equipment company. Microcentrifuges are widely used in biochemical laboratories and one can sometimes find a “retired” one there.
Others use syringe filters. We have avoided using syringe filters (aside from the fact that we have a microcentrifuge) because of the cost of the filters (>$1.00 each; see also section 3.3.10) and because there can be some loss of sample, which is a problem for very small samples.
3.2.7 An extraction protocol [from C. Mouri and R. Laursen, “Identification and partial characterization of C-glycosylflavone markers in Asian plant dyes using liquid chromatography—tandem mass spectrometry,” J. Chromatogr., A 1218, 7325-7330 (2011)].
Extraction of dyed fiber specimens was accomplished by heating approximately 0.1–1 mg of fibers in 200 µL of a solution of pyridine/water/0.5 M oxalic acid in water (90:90:20) at 100 °C for 15 min (Note 1). The mixture was cooled to room temperature and the solution was transferred to a 1.7-mL polypropylene microcentrifuge tube by pipet. The fibers were washed with 30 µL of water and the washings were added to the extract. [With larger samples, the washing step can be omitted.] The mouth of the tube containing the extracts was covered by stretching a small piece of Parafilm over it, after which several (ca. 20) small holes were punched in it using a needle or straight pin (Note 2). The extract and washings were then cooled to <0 °C in dry ice and evaporated to dryness at room temperature in a vacuum desiccator using a mechanical pump over a period of about 3 – 6 h, depending on the number of samples being evaporated. The resulting residue was dissolved or resuspended in 50 µL of methanol/water (1:1), the mixture was centrifuged at 12,000 rpm, after which 30 µL of the supernatant was transferred to an insert in an autoinjector vial for analysis; 20 µL was injected onto the HPLC column.
In the foregoing procedure, only about 40% of the extract used for LC–DAD-MS analysis. In the case of certain extremely small museum samples, it was necessary to analyze as much of the extract as possible. In this case, after evaporation of the pyridine solution, the sample residue was mixed with 100 µL of methanol. This solution was centrifuged at 12,000 rpm for 8 min, and 80 µL of the supernatant was transferred to a new microcentrifuge vial. To the 20-µL residue containing any precipitate was added an additional 50 µL of methanol; this mixture was centrifuged, and 50 µL was removed and added to the 80 µL of original supernatant. The combined washes (∼130 µL) were evaporated to dryness, the residue was dissolved in 25 µL of methanol/water (1:1), and all of this solution was transferred to the insert of an autoinjector vial; 20 µL was injected. In this way, about 75% of the total extract could be used for analysis.
Note 1. In the original version of the above procedure, we used 10 µL of 1.0 M oxalic acid. However we found, after a few weeks, that crystals of oxalic acid had formed in the stock solution of 1.0 M oxalic acid in water, apparently because the original solution was supersaturated. Crystallization was avoided by diluting this solution to 0.5 M.
Note 2. The purpose of this operation was to prevent, should it occur, as sometimes happens in a high vacuum, expulsion or “bumping” of the entire sample out of the tube, resulting in loss of the sample and possible contamination of nearby samples. This is more likely to happen if the pressure is reduced too quickly. For this reason, it is a good practice to reduce the pressure to about 20 mm Hg, wait a few minutes, and then continue to reduce the pressure to its lowest value.
3.2.8 Extraction of plant material. Extraction of plant material is analogous to what dyers do in preparing their dyebaths: they heat the plant material in water. In our studies, we have always extracted plant material in methanol-water (1:1) at about 60 °C, which is slightly below the boiling point (64 °C) of methanol. The methanol is added because many organic compounds are more soluble in alcohols than water. A chelation agent is not needed.
3.3 Analysis of dyes by HPLC
Chromatography is always a compromise between sample size, speed of the analysis and resolution. In books on chromatography, this is referred to as the “chromatographer’s triangle.” In the case of dye analysis, the samples are usually pretty small, so it is generally a compromise between resolution and speed of analysis: to get high resolution of closely eluting peaks, one often has to decrease the flow rate; conversely, if one wants to do the analysis quickly, one may lose resolution of some component pairs. Resolution also depends on the particle size of the stationary phase, with small particles giving better separations (J.M. Miller, “Chromatography: Concepts and Contrasts,” 2nd ed., Wiley-Interscience, 2005) .
3.3.1 Definitions. Chromatography, like most disciplines, has its own jargon. Although the substances of interest here are dyes, the definitions used by chromatographers are generally applicable.
HPLC: High Performance Liquid Chromatography
Analyte: The substance being analyzed (dye molecules, in this instance)
Stationary phase: The material on which separation takes place. In the case of dye analysis, it is usually a reversed phase material, which is packed into a stainless-steel tube; this comprises the “column.”
Mobile phase: The solvent used to elute the analyte from the stationary phase; for reversed phase analysis of dyes, this is usually mixtures of water and an organic solvent.
Reversed phase: A material made by covalently binding molecules to the surface of a stationary phase so as to change its polarity. For dye analysis, the most common materials are polar silica gel beads to which long chain hydrophobic groups (e.g., octadecylsilyl, or ODS) have been attached. Thus, the packing in a C18 column is silica gel beads to which are bonded octadecylsilyl groups (Figure 3.6).

Fig. 3. 6. Schematic representation of C18 packing material: octadecyl groups bonded to silica beads
Note: if all of the silica OH groups have not been bonded to non-polar groups, the support may retain some polar properties. As a result, dyes that contain ionic groups (ammonium, carboxylate, sulfonate) may interact the OH groups and elute as broad, tailing peaks. The relative elution positions of these peaks may also vary greatly from column to column. To reduce such effects, some HPLC packings are advertised as being “capped,” i.e., free OH groups are blocked with non-polar groups (e.g., methylsilyl groups).
Elution: The process of displacing (“washing out”) an analyte (e.g., a dye molecule) from a stationary phase (e.g., ODS-silica) by a mobile phase (e.g., a mixture of water and acetonitrile). An elution profile is a display of the analytes detected after displacement from the stationary phase as a function of time.
3.3.2 Choice of HPLC solvent (mobile phase). The two most widely used solvents for separation of dyes by reversed phase HPLC are acetonitrile and methanol, both of which are adequate in terms of performance. It has been argued that methanol is preferable because it is less expensive, but considering all the factors that contribute to the cost of analyzing a dye sample (See Table 3.4 in section 3.3.10), the difference in cost of the mobile phase is insignificant.
In our work we have always used acetonitrile primarily because mixtures of acetonitrile with water have significantly lower viscosities than those of methanol. As can be seen in Figure 3.7, the viscosity of 40-60 % methanol increases to about 180% that of water alone. And because the backpressure is directly proportional to viscosity (see Figure 3.8), wear and tear on the HPLC column and the pump components (e.g., seals) increases. For example, a run that starts out at a pressure of 200 bar in pure water, would increase to about 360 bar (5200 psi) at 50% methanol.
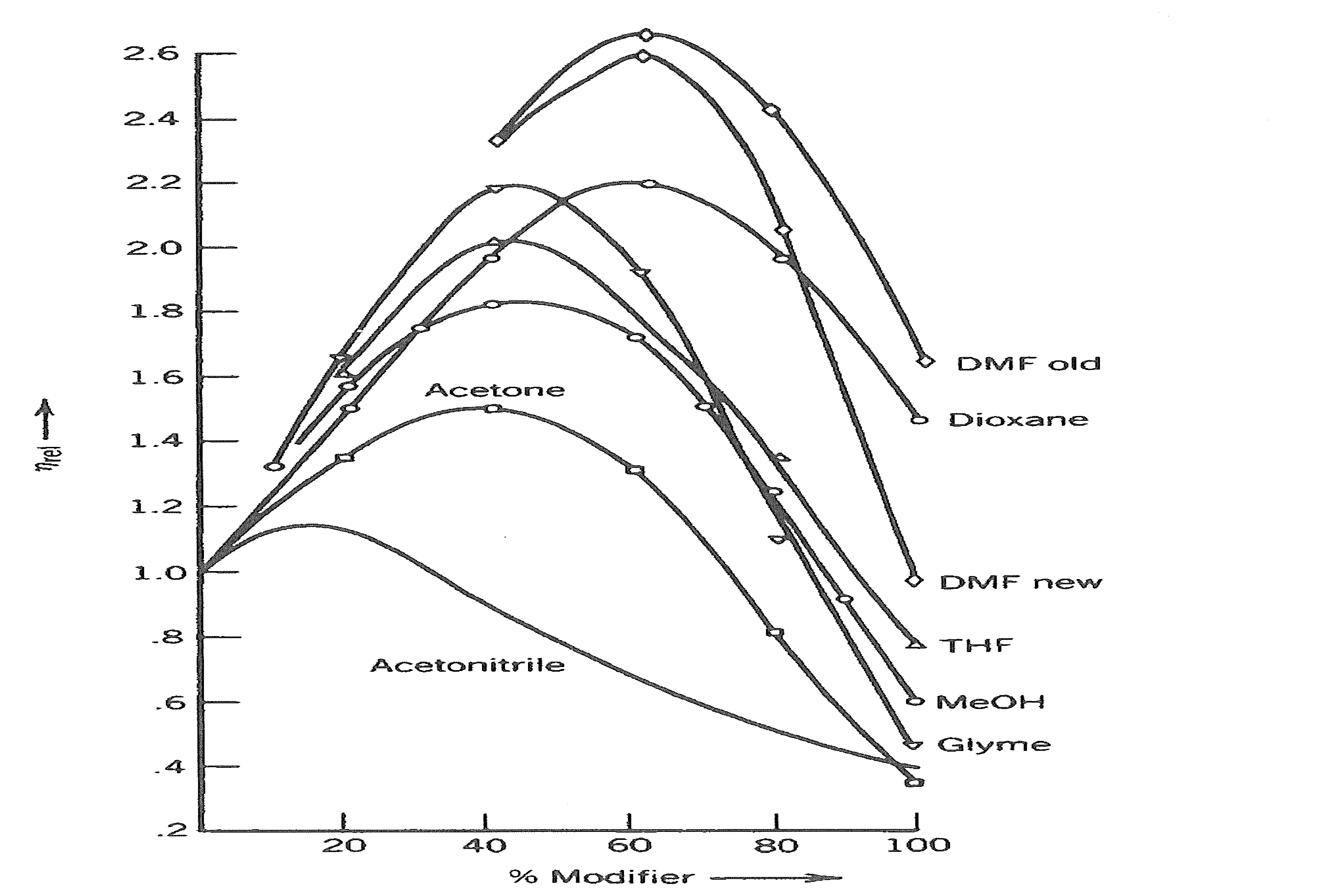
Figure 3.7. Variations in viscosity of water mixtures with increasing amount of solvents (From S. van der Waal, Chromatographia 1985, 20, 274, as reprinted in J.M. Miller, “Chromatography: Concepts and Contrasts,” 2nd ed., Wiley-Interscience, 2005, p. 238).
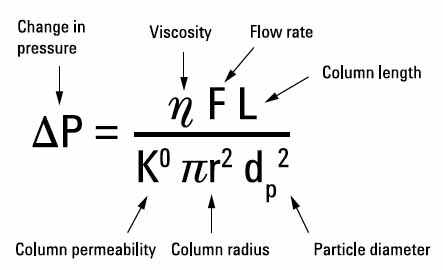
Figure 3.8. Back pressure as a function of various parameters (from Agilent Handbook)
3.3.3 Choice of column (stationary phase). Virtually all published HPLC separations of dyes have been done on reversed phase supports, which are generally silica gel beads, to which are bonded long hydrophobic molecules, most commonly, octadecylsilyl (ODS or C18) moieties. There are hundreds of types of column available. Most of them work more or less well, but they are generally expensive and only the most well-funded laboratory can afford to try many of them. As seen in Table 3.4 (section 3.3.10), the cost of the HPLC column is the greatest single consumable cost in the analysis of dyes. For some years we have used a 2.0-mm dia., 150 mm long, 3-µm particle size Phenomenex Luna column at a flow rate of 0.25 mL/min. No doubt other columns would work as well.
3.3.4 Loading the sample. Since dye analysis is generally done on reversed phase (e.g., C18 silica) columns, the sample should be loaded in as polar a solvent as possible—ideally water—so that sample forms a narrow (“tight”) band at the top of the column. Many dyes are poorly soluble in water but are fairly soluble in organic solvents. But samples cannot be loaded in pure organic solvents, either, because such solvents (e.g., acetonitrile and methanol) are often used to elute the sample (see below). Thus the sample may be loaded on the column as a broad band, which will result in its being eluted as a broad band, leading to poor sensitivity and resolution of dye components. For this reason, samples are often loaded in small volumes of a mixture of water and an organic solvent.
In our own work, using 2.1-mm dia. reversed phase columns, we have generally injected 20 µL of methanol-water (1:1), a solvent mixture that works for all dyes, although it dissolves only a small portion of indigo. Nevertheless, we always see detectable amounts of indigo (indigotin, indirubin, pseudoindirubin), even with this solvent. Other solvent mixtures and volumes probably could be used, too.
3.3.5 Elution. Elution of dye molecules from reversed phase columns is accomplished using gradients of water with some organic solvent, usually acetonitrile or methanol. The optimal gradient is dependent on many factors—particular HPLC column, solvent delivery system (pumps), specific separation required, elution time, etc.—and must be determined empirically depending on the system available and specific requirements. In general, the elution gradient ranges from 100% water (0% organic solvent) to about 95% organic solvent (5% water) over 15 – 90 min. We recommend not using 100% solvent because some dyes may precipitate under these conditions (on the other hand, it may not matter whether one goes to 100% or not). The gradient itself should be followed by 2 – 3 minutes (at a minimum) elution with 95% organic solvent so as to clean late-eluting components off the column. This is then followed by a short gradient to restore initial conditions and a few minutes of the initial solvent. Figure 3.8 shows some typical elution curve shapes.
Note: when using electrospray ionization (ESI) with a mass spectrometric detector, it is necessary to include an acid (usually 0.1% formic acid) in the elution solvents. The concentration should be the same in both solvents to avoid baseline shifts and to deliver a constant concentration of protons to the mass analyzer.

Figure 3.9 Typical elution profiles for a reversed phase HPLC column, eluted with water-organic solvent mixtures; %B indicates the percentage of organic solvent (usually acetonitrile or methanol).
The maximum elution flowrate depends on the diameter of the column and the particle size. We use 0.25 mL/min for a 2.0 mm dia., 150-mm long column packed with 3-µm particles and an overall run time of 36 min.
3.3.6 Column diameter. Assuming that one is using MS, as well as DAD, detection, it is an advantage to use a fairly small diameter column as this reduces, by a factor of 1/d2 (where d = column diameter), the volume of solvent entering the mass spectrometer. Since detection is generally done by electrospray ionization, the HPLC solvent has to be evaporated before analysis can occur. The less solvent there is, the less must be evaporated. As can be seen in Table 3.3, columns used with MS detection generally have diameters less than or equal to 2.1 mm. As discussed in the section 3.2.5 (Extraction volumes) one can gain sensitivity by using smaller diameter columns, but there is a loss in robustness.
Table 3.3. Analytical objectives (from Agilent Handbook)
| Comments | Column diameter (mm) |
|---|---|
| Very high sensitivity, LC/MS, peptides and proteins | 0.1, 0.075 |
| Very high sensitivity, limited sample, LC/MS, peptides and proteins | 0.3, 0.5 |
| High sensitivity, limited sample, LC/MS | 1.0 |
| Save solvent; special low-volume instrumentation is available | 2.1 |
| Special detectors such as MS | 2.1 |
| High sensitivity, limited sample | 2.1 |
| Save solvent; standard HPLC equipment available, LC/MS | 3.0 |
| Standard separations | 4.6 |
| Small-scale (mg) preparative separations | 9.4 |
| Medium-scale preparative separations (100 mg to g) or semi-prep | 21.2 |
| Large-lab-scale preparative separations (up to 100 g) | 30, 50 |
3.3.7 Particle size. In general, the smaller the particle size, the better the resolution. However, as can be seen from Figure 3.7, the backpressure of the system increases inversely with the square of the particle size. Thus, for the same size column and flow rate, the respective backpressures for particles of 5 µm, 3µm and 1.8 µm would be, relatively speaking, 100 bar, 280 bar and 770 bar (11,165 psi). To some extent, this increase in backpressure can be compensated for by reducing both the flow rate and the column length (see Figure 3.7).
3.3.8 Guard columns. Replacing a HPLC column can be very expensive, so it is advisable to take care of the one that one has. This can be done by loading clean samples onto it. Centrifugation or filtration can be used to remove particulate matter, which will otherwise accumulate on the top of a column and clog it, but this does not remove soluble polymeric or other “garbage” often found in extracts—especially plant extracts. Such “garbage” accumulates on the column and eventually reduces the performance (resolution) to the point that it has to be replaced. Use of a guard column is the most effective way to do this. A guard column usually utilizes a cartridge containing a small amount of the same/similar material found in the separation column. Thus the “garbage” (and any particulate matter) accumulates in the cartridge, which can be replaced periodically at relatively low cost (see Table 3.4).
3.3.9 Ion pairing. Ion pairing is a technique used for ionic species (e.g., direct dyes such as protoberberines and sulfonated synthetic dyes) that tend to give broad, tailing peaks on reversed phase columns. In this technique, an ion pairing substance (e.g., certain detergents) having both an ionic moiety that pairs with the ionic dye and a hydrophobic moiety that interacts with the hydrophobic reversed phase material of the column, is added to the mobile phase (e.g., acetonitrile and water). This results in sharp peaks for ionic substances.
There are, however, two serious problems with ion pairing: (1) it cannot be used with mass spectrometric detection because the ion pairing substance in the eluant is non-volatile and goes into the mass spectrometer resulting in extremely high background, and possible ruin of the detector, and (2) because of the strong hydrophobic binding between the pairing agent and the stationary phase, it is extremely difficult to remove the ion pairing agent from the HPLC column. As a result, the column cannot be used again for regular reversed phase chromatography. Usually a column used with a particular ion pairing reagent has to be dedicated to that method only.
3.3.10 Analytical cost analysis. The primary costs, aside from machine time, for analyzing dye extracts are the solvents and supplies associated with doing the chromatographic separation. As can be seen in Table 3.4, the greatest cost item, by far, for a single analysis is the HPLC column itself, followed by the vials used for loading the sample into the autoinjector. Some believe that using methanol as the elution solvent will save money because it is less expensive that acetonitrile, but as can be seen in Table 3.4, the cost difference is insignificant. Furthermore, as can be seen in Figure 3.6, methanol-water mixtures can result in an 80% increase in the back pressure on the column, possibly decreasing the lifetime of the column and components (e.g., seals) of the HPLC system.
Table 3.4. Approximate costs of consumables used for dye analysis (Jan. 2015 prices)
| Item | Bulk unit (Vol. or no.) | Cost/unit ($US) | Vol. or no. used/analysis | Cost/analysis ($US) |
|---|---|---|---|---|
| HPLC | ||||
| Acetonitrile (LCMS)* | 4 L | $150 | 5 mL | $0.19 |
| [Methanol (LCMS)* | 4 L | $130 | 5 mL | $0.16 |
| HPLC column (@1000 runs/col.) | $700 | 1/1000 | $0.70 | |
| Guard column cartridges (@500 runs/cartridge) | 3 | $180 | 1/500 | $0.12 |
| Sample preparation | ||||
| Microcentrifuge tube (PP) | 5000 | $105 | 2 | $0.04 |
| Pipet tips (200 and 1000) | 960 | $31 | 2 | $0.07 |
| Vial** (#408213) | 100 | $15 | 1 | $0.17 |
| Cap** (#531008) | 1000 | $40 | 1 | $0.04 |
| Septum** (#400581) | 1000 | $121 | 1 | $0.12 |
| Insert (polypropylene with spring)** (#CTI-2510) | 100 | $19 | 1 | $0.19 |
| Total (using acetonitrile) | $1.64 |
* Pharmco-Aaper (http://www.pharmcoaaper.com/pages/Products/chemicals.html)
Only one solvent is needed. Both are listed for price comparison.
**ChromTech, Inc. ( www.chromtech.com)
3.4 Instrumentation
There are many types and manufacturers of HPLC and detection systems, but the most useful for dye analysis is one the combines an HPLC separation system (preferably with automated loading) with tandem diode array (DAD) and mass spectrometric (MSD) detectors. With this sort of system, one can directly correlate peaks detected by DAD with their molecular masses. Nearly all of the data here were obtained using a system consisting of an Agilent 1100 HPLC system with automated sample loading, an Agilent 1100 diode array detector and an Agilent VL-API-ESI (electrospray), single quadrupole mass detector. Our system is nearly 20 years old, and many newer systems are now available that have higher sensitivity, better analytical software, and better capabilities for mass ion analysis (e.g., simultaneous detection of positive and negative ions and analysis of fragments, as with ion trap detectors). In general, though, fragment ion analysis (higher than MS2) is not helpful.
The diode array detector allows one to detect dyes of a particular color(s), and to record absorption spectra for each peak, but since many dyes (e.g., flavonol glycosides) have almost identical spectra, these spectra allow one only to place the dye in a certain group. However, if one can also determine the mass of the compound, one can eliminate a lot of possibilities. On the other hand, measure of the mass alone does not tell the analyst what color the compound is—of whether it has any visible color at all.